


Metals, Amyloid Beta and Alzheimer's Disease
Let's start with aluminium

As many of you are already aware the peptide known as amyloid beta is considered a central player in the aetiology of Alzheimer’s disease. It holds this position primarily through its presence as something called a senile or neuritic plaque in brain tissue in Alzheimer’s disease. However, counter arguments identify that not all sufferers of Alzheimer’s disease have senile plaques in their brain tissue and many aged individuals with senile plaques in their brain tissue do not have Alzheimer’s disease. A further anomaly concerns the fact that while it is irrefutable that amyloid beta is found in human brain tissue its concentration in body fluids never reaches the solubility product of the peptide. So, the latter means that it should not precipitate in brain tissue, it should be present in the extracellular fluids of the brain as the soluble, monomeric peptide. But it is found in brain tissue as insoluble aggregates and so something must be catalysing the precipitation of amyloid beta in brain tissue.
Perhaps metal ions are the catalysts and just perhaps this is why metals are found co-deposited with amyloid beta in senile plaques in Alzheimer’s disease. Before considering this possibility in detail it is also important to know that the forms of amyloid beta found deposited in brain tissue in Alzheimer’s disease (and in healthy brains) are not all the same. When amyloid beta is found at the core of senile plaques it is found in what is known as a beta sheet conformation, hence why it is called amyloid beta. Beta sheets are generally aberrant forms of protein and additionally may have greater stability being more difficult to break up by protease enzymes and more difficult to digest/process by macrophages such as the microglia in brain tissue. This enhanced stability may explain why senile plaques exist in brain tissue and why they have not been removed as would normally be the case for extracellular deposits of protein.
One metal consistently found co-deposited with amyloid beta in senile plaques in Alzheimer’s disease is aluminium. Of course, the aluminium industry along with their stooges in academia have desperately tried to explain the presence of aluminium in senile plaques as contamination, not of brain tissue, but emanating from reagents and processes used to image and quantify metals (aluminium) in human brain tissue. Our research over the past 12 years or so does now at least seem to have shut them up. Perhaps the piece de resistance being our recent research on aluminium in brain tissue in familial Alzheimer's disease. Of equal importance to the presence of aluminium in senile plaques is our observation way back in 1993 that amyloid beta monomer binds aluminium and in doing so the monomeric amyloid beta is changed into a beta sheet structure, the exact form that amyloid beta is found in senile plaques in Alzheimer’s disease. We published this seminal piece of information over 30 years ago, one might have thought it would have had an impact in Alzheimer’s disease research by now.
Just over 10 years ago I wrote a paper reviewing mechanisms whereby amyloid beta bound aluminium and precipitated as senile plaques. I have copied the figure from this paper below and followed it with some explanatory text for those of you who like to get down to the details.
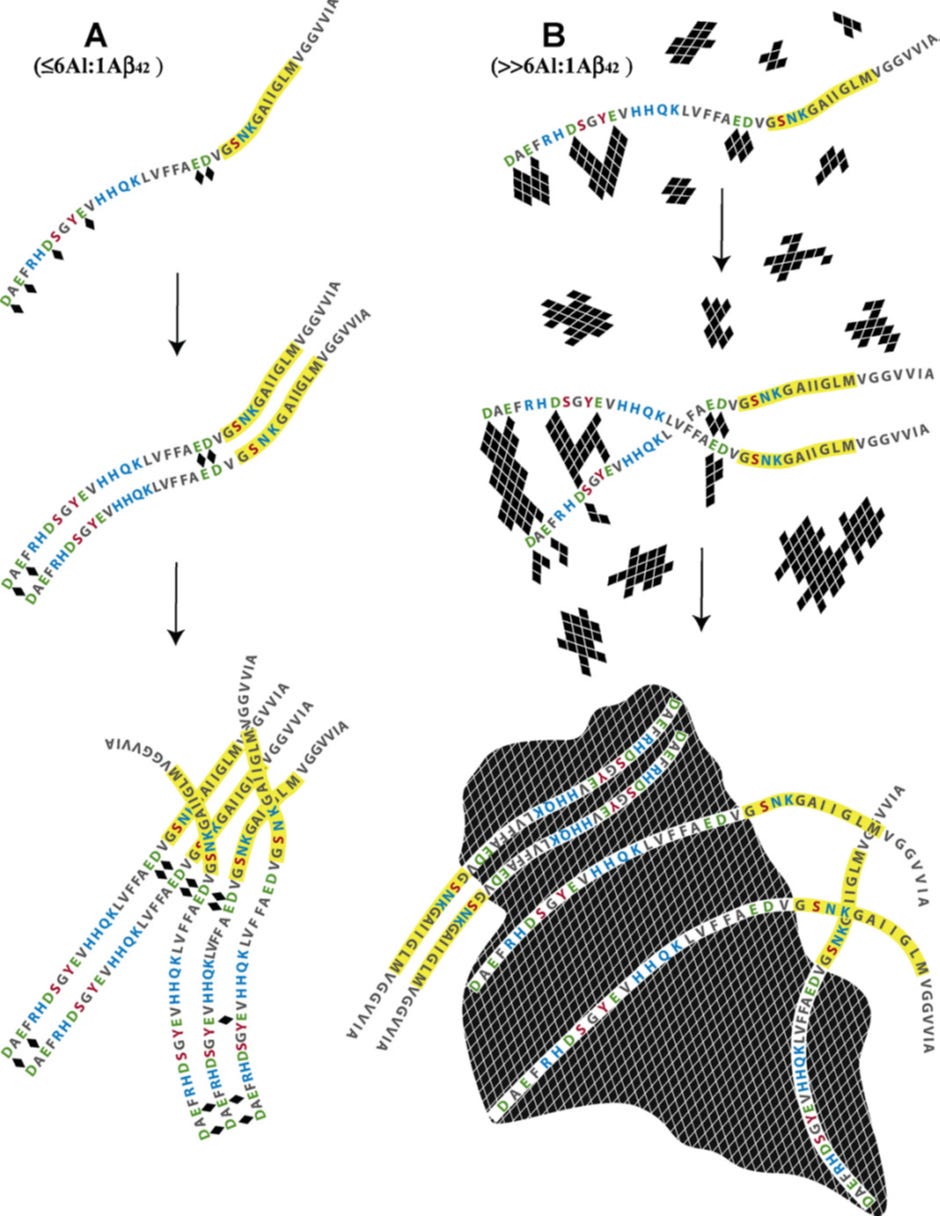
Schematic describing the potential mechanisms whereby aluminium, as Al3+(aq) and Al(OH)3(s), interacts with Abeta42 (numbered 1–42 from left to right) to both promote (A) and delay (B) the peptide’s re-arrangement into beta sheet structures characteristic of senile plaques in vitro and in vivo.
(Image A) The coordination of aluminium (indicated above as ♦), as Al3+(aq), by Abeta42 is most likely to involve carboxylate functional groups associated with the negatively charged (under physiological conditions) amino acids glutamate (Glu, E) and aspartate (Asp, D). Asp1,7,23 and Glu3,11,22 are all possible binding sites with additional stability of any subsequent Abeta42–Aln complex being offered by adjacent polar amino acids such as arginine (Arg, R), histidine (His, H), serine (Ser, S), tyrosine (Tyr, Y), glutamine (Gln, Q) lysine (Lys, K) and asparagine (Asn, N). Experimental evidence has predicted that Abeta42 binds up to 6 Al3+(aq) giving a maximum stoichiometric ratio of 6Al:1Abeta42. Under dilute conditions (≤6Al:1Abeta42) Al3+(aq) is bound by D/E on adjacent peptides to form links or Al-bridges which under certain conditions will bring the highlighted region (Gly25–Met35, which includes the nucleation site for acquisition of beta sheet structure) of each peptide into closer proximity and thereby potentially to promote amyloidogenesis. The Glu22/Asp23 couplet should be a preferred binding site for Al3+(aq) with the subsequent formation of Al-bridges being capable of promoting the formation of beta sheets of amyloid. The observation that copper, in competition with aluminium, prevents Abeta42 from forming beta sheets through copper being bound by His6, 13/14 and Asp1 might also implicate Al3+(aq) binding by Asp1 and/or Glu3 in aluminium-promoted amyloidogenesis.
(Image B) Aluminium is only sparingly soluble under physiological conditions and the concentration of Al3+(aq), the biologically-reactive form of aluminium, will be determined by hydrolytic equilibria which favour both Al(OH)3(s) and Al(OH)4−(aq). Under conditions where aluminium is in significant excess to the peptide (>6Al:1Abeta42) Abeta42 upon binding Al3+(aq) will become occluded within rapidly forming aggregates of Al(OH)3(s) and thereafter will be co-precipitated with the solid phase. During the early stages of this process, amyloidogenesis will still be possible though with the rapid increases in both number and size of the aggregates of Al(OH)3(s) the opportunities to bring together the amyloid-nucleating regions of the occluded Abeta42 peptides will diminish equally as rapidly. The Abeta42 peptides are ‘harvested’ from solution by the rapidly hydrolysing Al(OH)3(s) in much the same way as natural organic matter is removed in the clarification of potable waters by aluminium salts.
The research that my group and others has published over decades now provides unequivocal evidence that aluminium co-deposits with amyloid beta in Alzheimer’s disease. Why does a normal physiologically important peptide precipitate as beta sheets in brain tissue in Alzheimer’s disease. This feels wrong and goes against what one might expect from an evolutionarily conserved mechanism in brain biochemistry. Of course, aluminium is a newcomer to brain biochemistry and this may be the key to the appearance of this aberrant process in Alzheimer’s disease.
In a subsequent blog I will discuss how different the case is with the essential metal copper.
Regardless of anything else, the spraying of nanoparticulate aluminum, barium, and strontium in the atmosphere should be terminated by any method required.
geoengineeringwatch.org
As usual, excellent information! Based on your book and posts, I recently ran a Doctor's Data UTM study. https://www.doctorsdata.com/Urine-Toxic-and-Essential-Elements I ran a blood test for aluminum and it the results were mid-range at 5 which is of concern. As such, I went looking for aluminum in my urine but found that I am at 75th percentile for LEAD! Surprisingly, my aluminum was low at 3.2 u/g.
Per my NMD, lead also causes cognitive issues similar to AD. I am now searching for where the lead is coming from? A few weeks ago, I dropped off a total of 6 samples at a local analytical lab for full ICP scans. So far, I have found a little aluminum and very little lead which means I will be dropping more samples off to the lab after another discussion with my NMD.
Have you seen any lead in brain tissue while working on aluminum?